FDA issues electronic trial data guidance to support accuracy
Increased computer use in clinical trials creates problems because data can be easily copied, transferred, changed or deleted without obvious evidence. To help ensure accurate, original and attributable data is available for clinical review and site inspection guidance has been issued.
The US Food and Drug Administration (FDA) draft guidance breaks handling electronic clinical trial data down into three phases: entry; review; and processing and transmission. An FDA chart showing how data might flow between the three phases is pictured below.
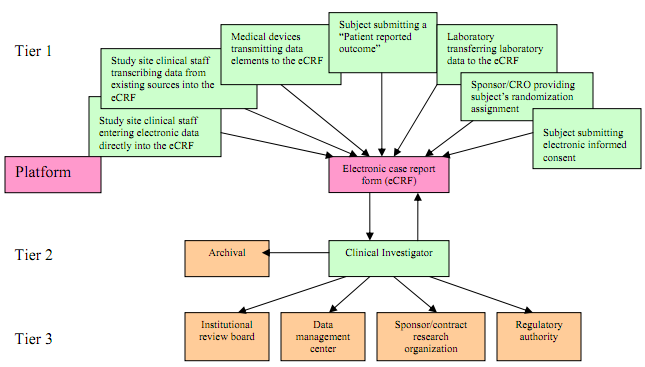
Data entry is the focus of the majority of the guidance. In the guidance the FDA identifies two types of data: information generated at a study visit and entered directly into electronic case report forms (eCFR); and elements transcribed from a source document, such as a lab report.
When looking into information entered directly into eCRFs the FDA may request documents to corroborate the data. For transcribed data the FDA says source documents should be kept and made available, if requested, to inspectors.
Complying with these, and a number of other, recommendations in the guidance should help ensure data is traceable. These measures ensure electronic information can be “traced through complex data capture, transmission and archival processes” for regulatory purposes.
Data element identifiers are part of creating an audit trail. These should identify: the originators of the information, including those entered manually by an investigator and automatically by a device; the date and time of submission; and the study subject referred to by the data.
Comments and suggestions regarding the draft should be submitted to the FDA by April 7.