Promotional Features


The role of dissolution in drug development
By Steve Alley, Pharmaceutical Development Project Manager
Oral dosage forms remain one of the most flexible and effective treatments available to patients. Dissolution testing is a requirement for all solid oral dosage forms and is used throughout the development life-cycle for product release and stability testing. It is a pivotal analytical test used for detecting physical changes in an active pharmaceutical ingredient and formulated product. At the early stages of the drug development process, in-vitro dissolution testing underpins the optimisation of drug-release from a given formulation.
The effectiveness of an oral dosage form depends upon the intrinsic ability of the drug to dissolve in the fluids of the gastrointestinal tract prior to being absorbed into the circulation. Therefore, the rate of dissolution of the tablet or capsule is pivotal to this process.
One of the key issues encountered within pharmaceutical development is the need to optimise the level of drug available to the body in order to have its desired therapeutic effect. Incorrect optimisation of this outside the therapeutic window could potentially lead to insufficient bioavailability (and hence lack of efficacy) or conversely, too high a bioavailability, producing unwanted adverse toxic effects to the patient.
In order to assist with dosage form optimisation, dissolution testing is a standardised method for measuring the rate of drug release from a given dosage form.
Despite being a commonly employed test in the pharmaceutical and biopharmaceutical industry, the fundamentals of dissolution testing are very often not correctly understood. It is imperative that the test is both robust and reproducible, with the ability to detect any key changes in product performance i.e. discriminate between different formulations and/or batches.
The exact dissolution technique employed is determined by the dosage form characteristics and the intended route of administration. For solid dosage forms, the industry standard dissolution testing methodologies are the United States Pharmacopoeia (USP) Apparatus I (basket) and USP Apparatus 2 (paddle). Immediate, modified and extended release are usually tested in standard dissolution baths with USP 2 paddles. For oral dosage forms that are prone to floating, USP 1 (baskets) would generally be required. Apart from the aforementioned apparatus types, there are also other techniques available such as USP 3 (reciprocating cylinders), USP 4 (flow-through-cell), USP 5 (paddle-over-disc), USP 6 (cylinder) and USP 7 (reciprocating holders).
In terms of developing a dissolution procedure, there are several aspects that need to be evaluated in order to achieve a robust method. Factors such as apparatus type, media and agitation rate must all be evaluated, and should be appropriate for the product being tested. For the purposes of this article, the most commonplace techniques (USP apparatus 1 and 2) will be discussed, including a summary of typical method parameters that should be considered when developing a dissolution method.
For a dosage form to be efficacious, the API must be both extracted (dissolved in solution) and then absorbed into the systemic circulation to facilitate its transport to the active site. This process influences the overall bioavailability of the API. Understanding this process is fundamental in undertaking in-vitro method development. Dissolution is the process of extracting the API from the solid dosage form into solution. A simple diagrammatic representation is given below:
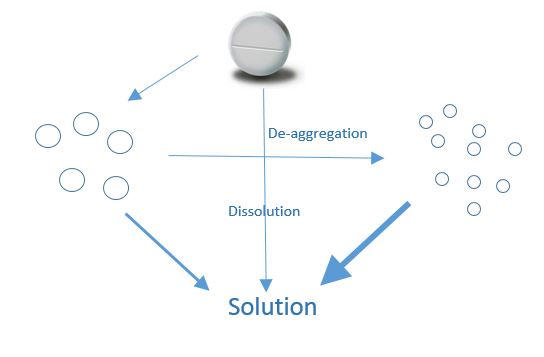
Fig 1: Dissolution of a solid oral dosage form
Once the API is in solution, the process of absorption can take place, whereby the drug substance is passed from the gastrointestinal lumen into the circulatory system where it can then travel to the relevant receptor sites to exert a biological response.
Dissolution is an in-vitro test that evaluates how an API is extracted from a solid dosage form. It can provide an indication of the efficiency of in-vivo dissolution but by no means provides any data on drug substance absorption. In order to assess the latter, pharmacokinetic data would need to be generated and used to supplement the dissolution work.
If appropriate in-vitro conditions are selected and appropriately evaluated (to simulate in-vivo conditions), it can aid in determining successful in vitro-in vivo correlation (IVIVC), or, worst case, a relationship between the two. It should be noted that dissolution parameters that are optimal for QC purposes may not be suitable for establishing IVIVC, so two dissolution tests may well be required to satisfy both development and regulatory needs.
When developing a dissolution method, it is important to take a logical, systematic approach to the process, and ensure that both the scientific and regulatory principles are borne in mind. A robust dissolution method should be free of significant interferences (e.g. matrix effects due to excipients), give low variability (precision) and produce a good profile shape. The method should also be challenged to discriminate between batches of material with different quality attributes. Once the process of identifying suitable medium and apparatus are complete, further optimisation of the method would be required to evaluate ionic strength of the medium, agitation rate and, if required, surfactant concentration. The final developed method should have the ability to discriminate between different formulations/batches, but still maintain acceptable precision and robustness. With regards to precision, typical limits for early and later time-points would be <20% and <10% RSD respectively.

Fig 2: Typical dissolution bath apparatus
Sink conditions
Sink conditions are defined as ‘the solution concentration corresponding to typically 5-10 times the nominal working concentration of the API in the dissolution medium’. Confirmation of achievement of sink is critical in establishing a suitable dissolution method. If these are not able to be achieved (and hence the media reaches saturation point), the dissolution rate cannot be consistently measured. It is important that when conducting dissolution testing, the only influences on the result should be the agitation rate and solubility of the product.
Media
The initial focus when screening potential media is to start with those which are aqueous based, within the pH range of 1.2-6.8 at the recommended ionic strength (as per USP/EP). When assessing APIs that display low solubilities in aqueous media throughout the pH range, incorporation of a surfactant is advisable, although the levels used should be as low as possible.
Dosage form
The fundamental characteristics of the dosage form type (capsule, tablet etc.), strength, excipients, release type (immediate, sustained, delayed), stability data and surface coatings, should all be considered during the method development phase. Additionally, manufacturing variables can also be pivotal in evaluating differences in API release between different formulations. A well-developed dissolution method should allow discrimination of the various product attributes.
When performing a dissolution, samples would be removed at set time-points, and the percent dissolved against time plotted (as demonstrated in figure 3 below):

Fig 3: Dissolution profile showing discrimination between different formulations
Throughout the duration of the dissolution analysis, it is critical to ensure visual observations are maintained. This can provide additional data to support any quantitative differences in the analytical results.
The analytical finish for the dissolution needs to be established. Where possible, in order to maintain simplicity and efficiency (and assuming the presence of a suitable chromophore), a simple UV finish could be employed; this would also require suitable specificity to be obtained to ensure that the absorbance of the active is not compromised by any other interferences.
More often than not, quantitation would be performed using HPLC-UV. This can often be favoured in order to reduce/eliminate interferences that may otherwise compromise a stand-alone UV method. Additionally, it is very likely that a previous HPLC assay method can be adapted for the dissolution samples by curtailing the run-time to allow the high sample throughput required for dissolution analysis. HPLC also provides flexibility in adjustment of injection volume to accommodate variations in sample concentration that are likely to be needed for different product strengths.
There are occasions where it maybe a practical alternative to screen for elemental components of the material, for example for a product where the active material is an inorganic salt, or in situations where traditional detection is problematic, then it may be possible to monitor a counter ion such as sodium or calcium instead.
There are a variety of elemental spectroscopy techniques that can be employed for this purpose. The choice of instrument is dependent of the analyte required and the expected concentration in the solutions.
For example, a potassium bromide tablet dissolution profile could be obtained by use of either flame atomic absorption spectrometry (FAAS) to measure the level of potassium or alternatively the concentration of bromide (measured as elemental bromine) could be measured by inductively coupled plasma – mass spectrometry (ICP-MS). Another application may be the use of inductively coupled plasma – optical emission spectroscopy (ICP-OES) to ascertain the content of iron in solutions drawn from a ferrous fumarate tablet dissolution
In summary, the design of a suitable dissolution procedure must take into consideration many parameters spanning the API, formulation and analytical method. In-vitro dissolution testing should provide a robust body of data in order to assure product performance and quality. Throughout the process it is important to ensure that the in-vitro dissolution resembles in-vivo conditions. If the dissolution procedure is designed effectively, it will accelerate drug development, and ideally reduce the need for human studies.




