Promotional Features


Extractables and leachables analysis of pharmaceutical products
By Alan Wood, Reading Scientific Services Ltd - Pharmaceutical Analyst (LC-MS)
Introduction
A crucial undertaking when releasing pharmaceutical products for the market, is to determine the purity of the final product, necessitating the need to determine its impurity profile. Traditionally, this was concerned only with those impurities arising from the manufacture and degradation of the pharmaceutical product. Nowadays, migration of mobile chemical species from components used in the manufacture and storage of pharmaceutical products must be assessed.
Regulatory bodies such as the US Food and Drug Administration (FDA) and The European Medicines Agency (EMA) are increasingly focussing on the interactions between various manufacturing components, drug delivery devices and container-closure systems (CCS), and the final pharmaceutical product. The aim is to identify and assess any toxicological risks which could arise via such interactions.
The FDA has previously stated that “Drug product containers and closures shall not be reactive, additive, or absorptive so as to alter the safety, identity, strength, quality or purity of the drug beyond the official or established requirements.”1 Consequently, extractables and leachables (E&L) studies are now a crucial component of product release.
Extractables and leachables: definitions and sources
The FDA defines extractables and leachables as follows:2
Extractables: Organic and inorganic chemical species that can be released from the surfaces of components used in the manufacture and storage of drug products under laboratory conditions (accelerated or exaggerated temperatures, solvents or surface exposure).
Leachables: Organic and inorganic chemical species that can be released from the surfaces of components used in the manufacture and storage of drug products under conditions of normal use.
Extractables represent the worst-case scenario regarding release of mobile chemical species from manufacturing and packaging components during forced extraction. Leachables should then comprise a sub‑set within this pool of mobile chemical species, released under the gentler conditions of on-shelf storage.
In practice, however, the leachables study identifies species that were not all observed during the preceding extractables study. Thus, the set of leachable species is not wholly included within the set of extractables, but there is strong overlap between both sets (figure 1).
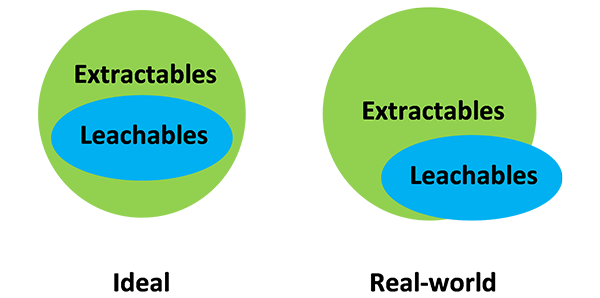
Figure 1: Relationships between extractables and leachables
There are multiple sources which can contain leachable species, and some common examples are detailed below:
- Manufacturing
- Filters
- Single-use bags
- Tubing
- Primary packaging - External components
- Container-closure system (vials, caps, lids and foils)
- Single-use systems (syringes and intravenous bags)
- Primary packaging - Internal components
- Gaskets and O-rings
- Valves
- Springs
- Secondary and tertiary packaging
- Labels and adhesives
- Inks and colourings
Leachable species to consider as part of an E&L study include:
- Antioxidants and stabilisers
- Anti-static coatings
- Lubricants, slip agents and emulsifiers
- Dyes and colourants
- Vulcanising agents
- Residual monomer, polymer and oligomer species
- Phthalates, nitrosamines and polyaromatic hydrocarbons (PAHs)
- Toxic elements – e.g. mercury, lead, arsenic, cadmium
The plethora of component parts used during product manufacture and storage, combined with the potential number of leachables, necessitates a structured and thorough E&L study.
E&L studies – a route to toxicological evaluation
Leachables that migrate into pharmaceutical products from the manufacturing and packaging system are required to be identified and monitored over the shelf-life of the product. The collated data permits a toxicological assessment to be made with respect to any leachables found, ensuring patient safety.
Key to this is determining the Safety Concern Threshold (SCT) for the product under investigation. SCTs were introduced by the Product Quality Research Institute (PQRI) leachables and extractables working group, and are defined as the dose below which an individual leachable would not elicit effects, carcinogenic or otherwise, which could constitute a safety concern.3
Two factors which impact significantly on the SCT are the route of administration and the degree of product-packaging contact. To assign risk from leachable species to the pharmaceutical product under investigation, the FDA developed the following matrix:4
Degree of concern associated with the route of administration | Likelihood of packaging component-dosage form interaction | ||
High | Medium | Low | |
Highest | Inhalation aerosols and solutions; injections and injectable suspensions | Sterile powders and powders for injection; inhalation powders |
|
High | Ophthalmic solutions and suspensions; transdermal onasal aerosols and sprays |
|
|
Low | Topical solutions and suspensions; topical and lingual aerosols; oral solutions and suspensions | Topical powders; oral powders | Oral tablets and oral (hard and soft gelatin) capsules |
Products such as aerosols and injectables have the highest associated risk, whilst oral tablets and capsules have the lowest risk.
The PQRI have recommended that the high risk SCT is set at 0.15 µg/day, whilst the low risk SCT is set at 1.5 µg/day, both having been justified from toxicological and safety perspectives.3
Under certain conditions, such as short-term exposure or in the treatment of a life‑threatening condition, the SCT can be raised above 1.5 µg/day.5
Designing an E&L study
An E&L study is composed of two individual yet interlinking projects. The extractables study identifies species from manufacturing components (where applicable) and the packaging system that could migrate into the pharmaceutical product upon storage under normal conditions. This establishes a baseline for the following leachables study, a series of tests carried out at predefined time-points on the pharmaceutical product for the duration of its shelf-life.
Extractables study
The components under investigation are extracted in isolation of the pharmaceutical product. Key points to consider are the number of components and material types that are to be tested, and the solvents with which to perform the extractions.
Simple storage systems, e.g. glass ampoules or plastic bottles with screw caps, will have a limited number of components. However, more intricate units, e.g. pump dispensers containing O-rings and springs, will contain multiple components which all require investigation. Secondary and tertiary packaging also needs to be considered at this stage.
Extractions should be performed with a range of solvents of varying solvating power, to ensure a representative pool of organic and inorganic extractable species are generated. In the case of liquid formulations, those chosen should best mimic the composition of the pharmaceutical product to provide a worst-case extractables profile. The use of overly-powerful solvents is discouraged. This could destroy component materials, creating an unrealistically large set of extractables.
Two or three solvents are typically chosen, but more can be used if considered appropriate. Common examples include:
- Water (neutral, and acidic or basic if pH ≠ 7)
- Organic solvent (ethanol, isopropanol or n-hexane)
One final aspect to consider is the material type under investigation. Plastics and rubbers should be extracted by all solvents chosen. However, there is little value in performing organic solvent extraction of metal springs, which would only yield inorganic impurities. Such considerations should be made between the solvents used and the components under investigation.
Leachables study
Time-points are predefined prior to commencing an E&L study, and can be run concomitantly with a stability trial.
Samples are screened for leachables, including those identified during the extractables study and any new species found during the leachables study. Those found to exceed the SCT are identified and assessed for toxicity.
Crucial to a successful study is the creation and storage of appropriate controls and leachables sample. The control sample should be stored in such a fashion that there is minimal risk of leachable ingress, and carefully labelled avoiding the use of inks and adhesives directly on the container. For the leachables samples, whether they should be stored inverted as well as upright (e.g. bottles fitted with caps or lids), and storage conditions (e.g. 4°C, 25°C/60% RH, 40°C/75% RH) should be considered.
Analysing extracts and samples
In general, extractables and leachables can be divided into three broad groups:
- Non-volatile leachables
- Volatile and semi-volatile leachables
- Inorganic / elemental leachables.
Validated analytical methods are required to analyse all samples, and can be used across both studies. Figure 2 exemplifies the typical analytical strategy employed.

Figure 2: Generalised approach to analysis of E&L samples
Liquid chromatography–mass spectrometry (LC-MS) analysis permits the analysis of larger, non-volatile leachables species. Direct injection gas chromatography mass spectrometry (GC-MS) analysis permits analysis of both volatile and semi-volatile leachable species. Head-space GC-MS analysis is an option where a great deal of volatile species may be expected. Elemental impurities are analysed and quantified by inductively coupled plasma mass spectrometry (ICP-MS).
Methods must be validated prior to commencing work, but can accommodate a wide range of pharmaceutical products. Typically, method feasibility needs only to be demonstrated prior to commencing leachables studies. This ensures that the pharmaceutical product under investigation does not elicit inhibitory matrix effects, and that extractables can be recovered from the sample matrix, avoiding method redevelopment and revalidation.
Conclusions
The complex nature of the E&L study requires thorough planning and access to a variety of complex analytical hardware running validated methods.
The investigative nature of the work demands a team of analysts with capabilities across method validation, molecular identification and toxicological evaluation, to ensure that the study is run smoothly and data is interpreted correctly.
With thorough planning, appropriate analytical hardware and in-depth knowledge in place from the beginning, the E&L study can be run smoothly and successfully to completion, ultimately ensuring that patient safety is maintained.
References
1. US FDA, Code of Federal Regulations, 21CFR211.94 - http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=211.94
2. Current FDA Perspective on Leachable Impurities in Parenteral and Ophthalmic Drug products, 2011 - https://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/UCM301045.pdf
3. Development of Safety Qualification Thresholds and Their Use in Orally Inhaled and Nasal Drug Product Evaluation - DOI: https://doi.org/10.1093/toxsci/kfm058
4. Guidance for industry: Container closure systems for packaging human drugs and biologics - http://www.fda.gov/downloads/Drugs/Guidances/ucm070551.pdf
5. Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk, M7 - http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M7/M7_Step_4.pdf




